Examples for GEPIA Usage
With GEPIA, experimental biologists can easily explore the TCGA and GTEx datasets, find answers for their questions, and test their hypotheses.
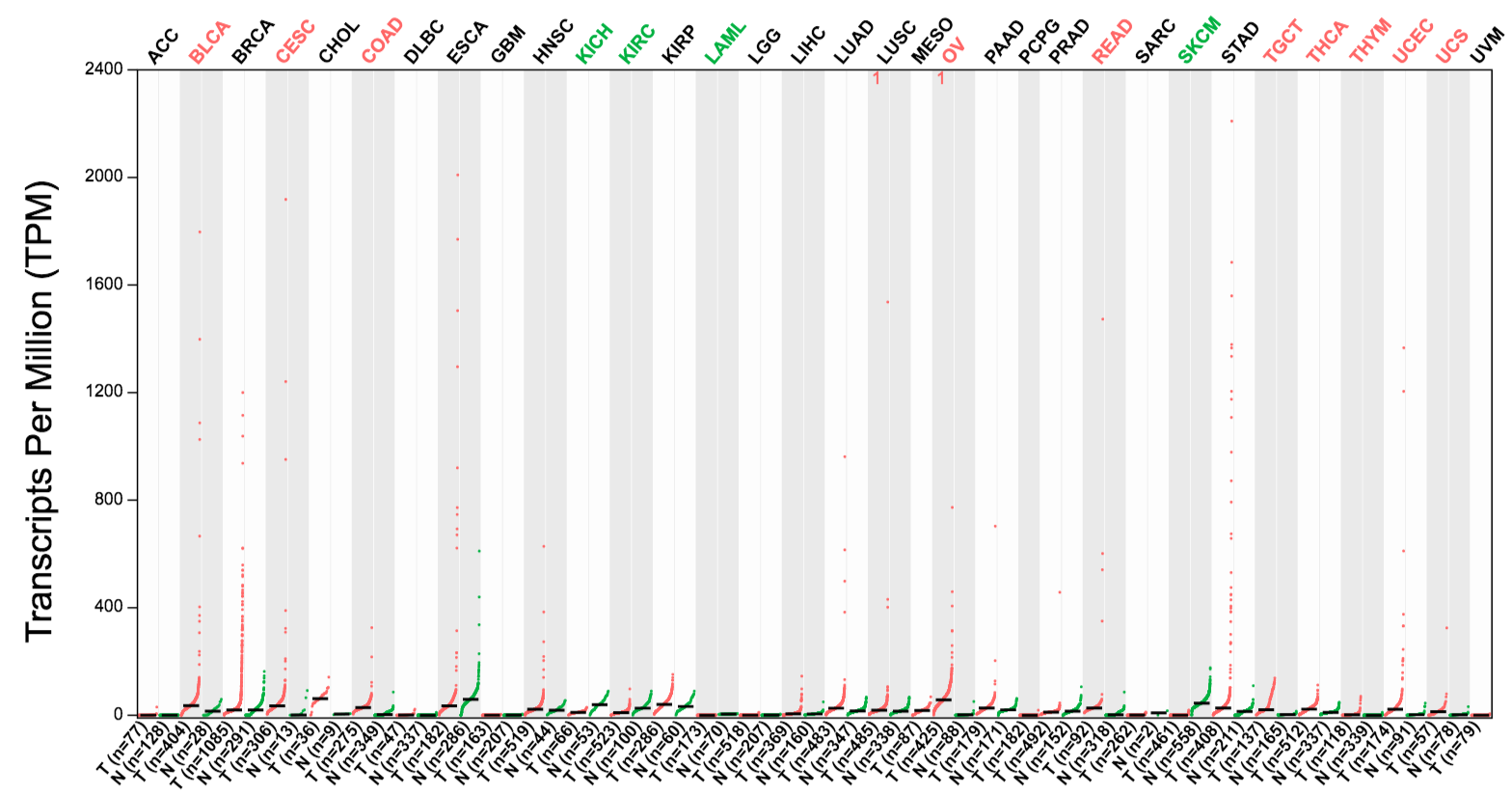
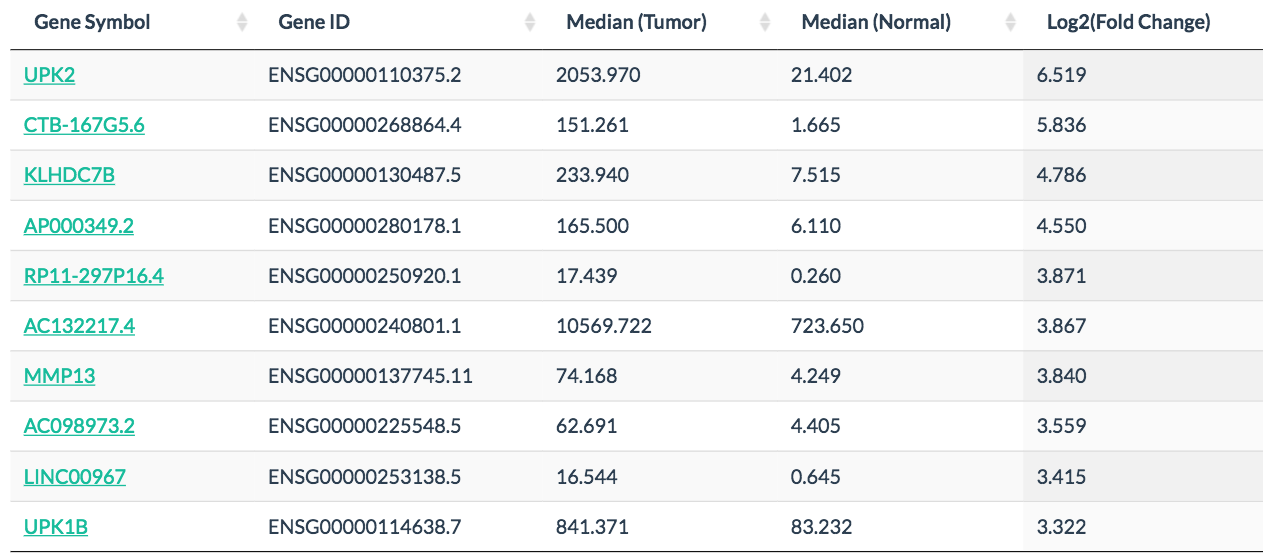
In differential analysis and expression profile, users can easily discover differentially expressed genes, such as MPO in leukemia and UPK2 in bladder cancer.
MPO specifically expressed in leukemia:
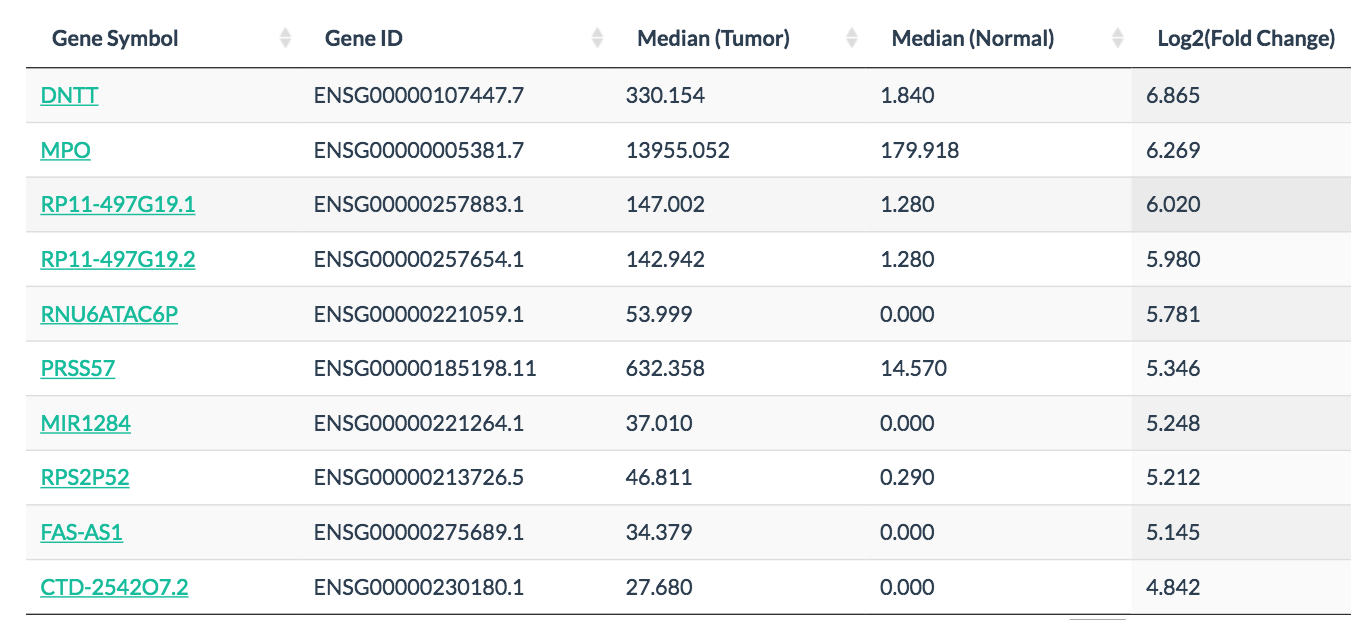
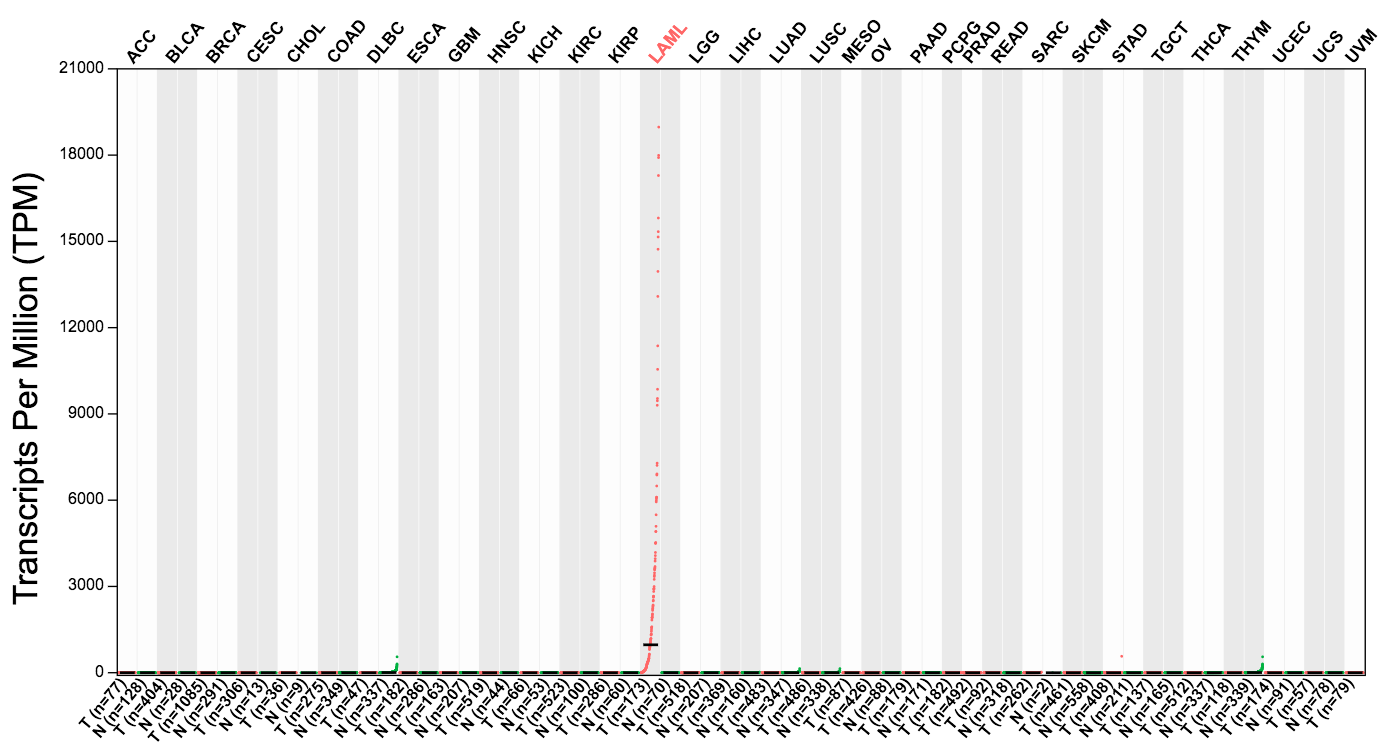
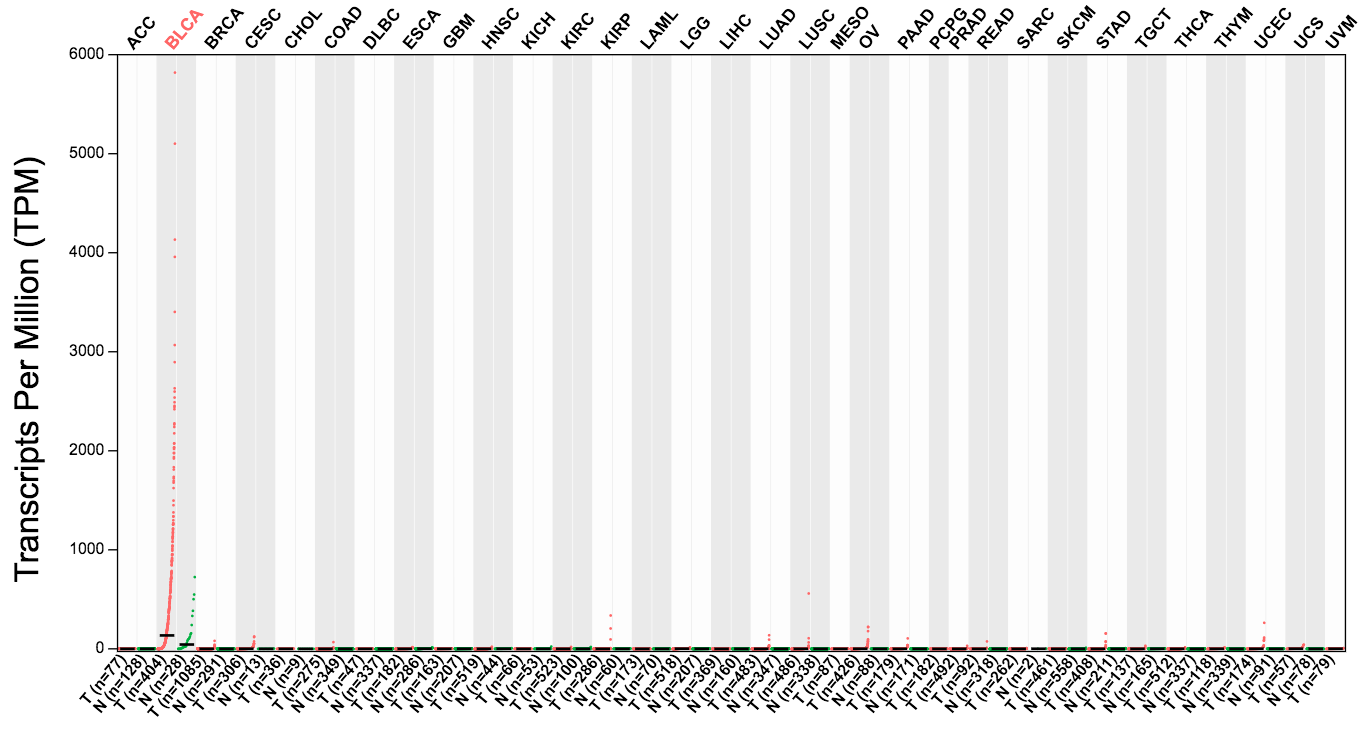
UPK2 specifically expressed in bladder cancer:
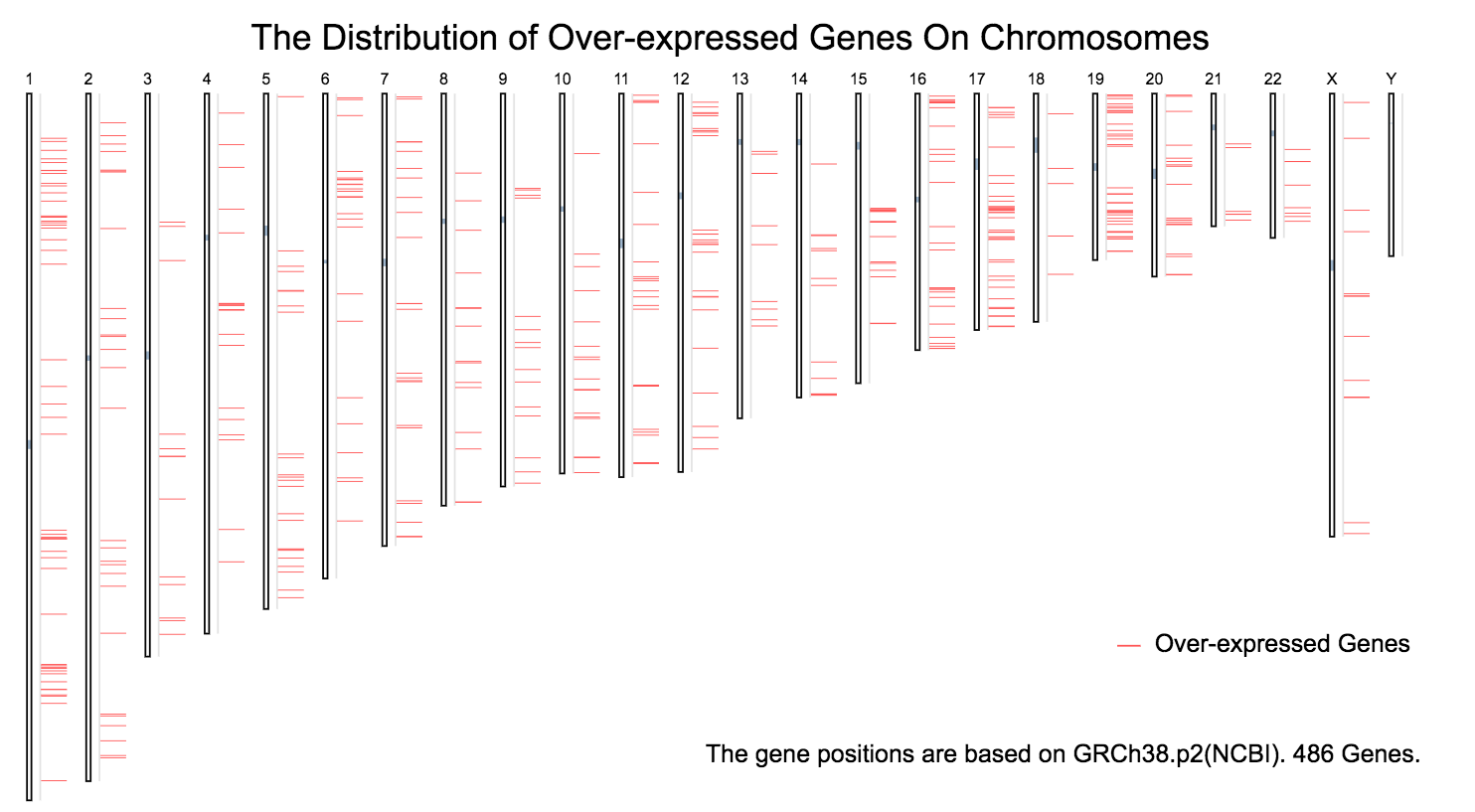
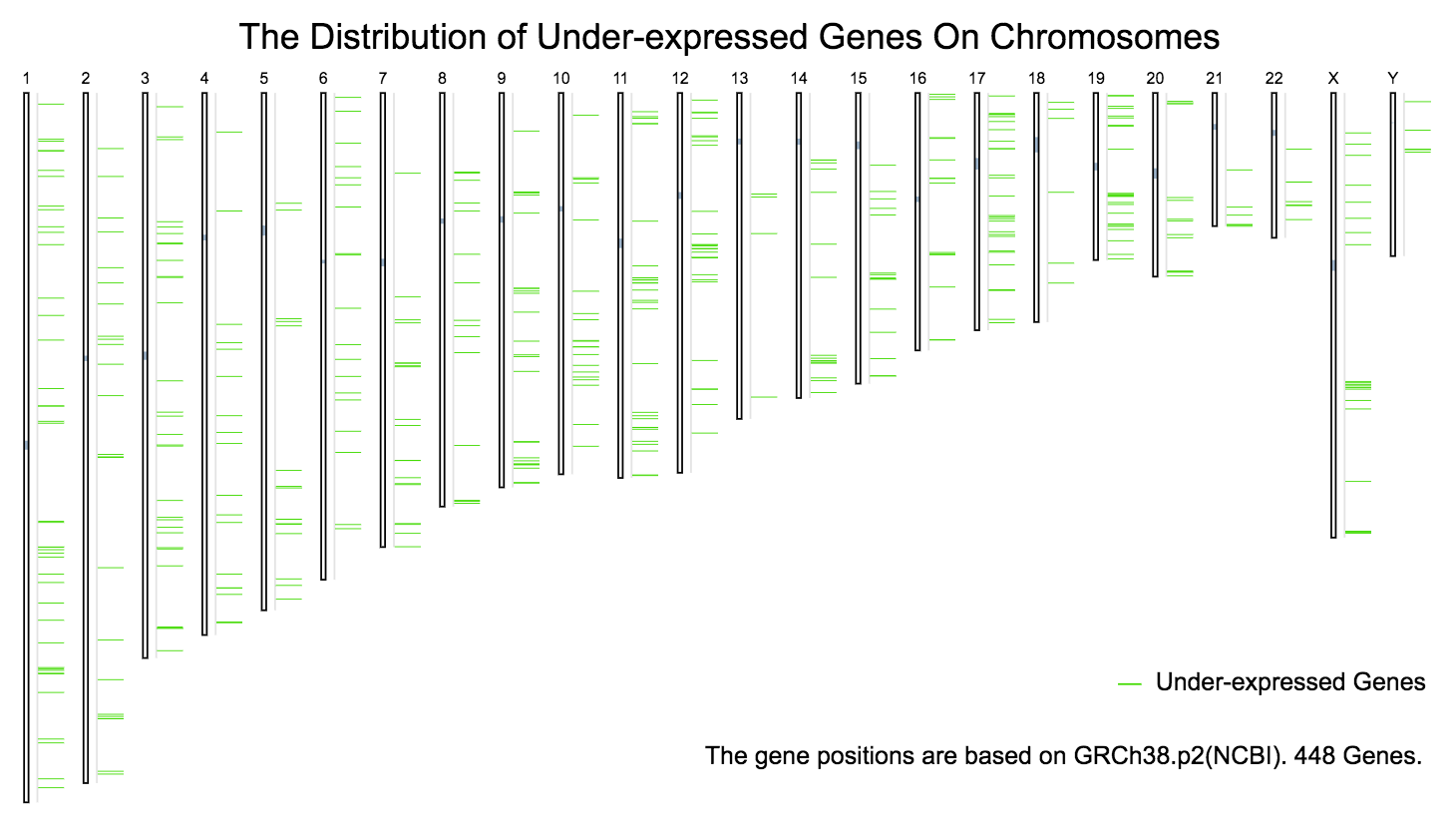
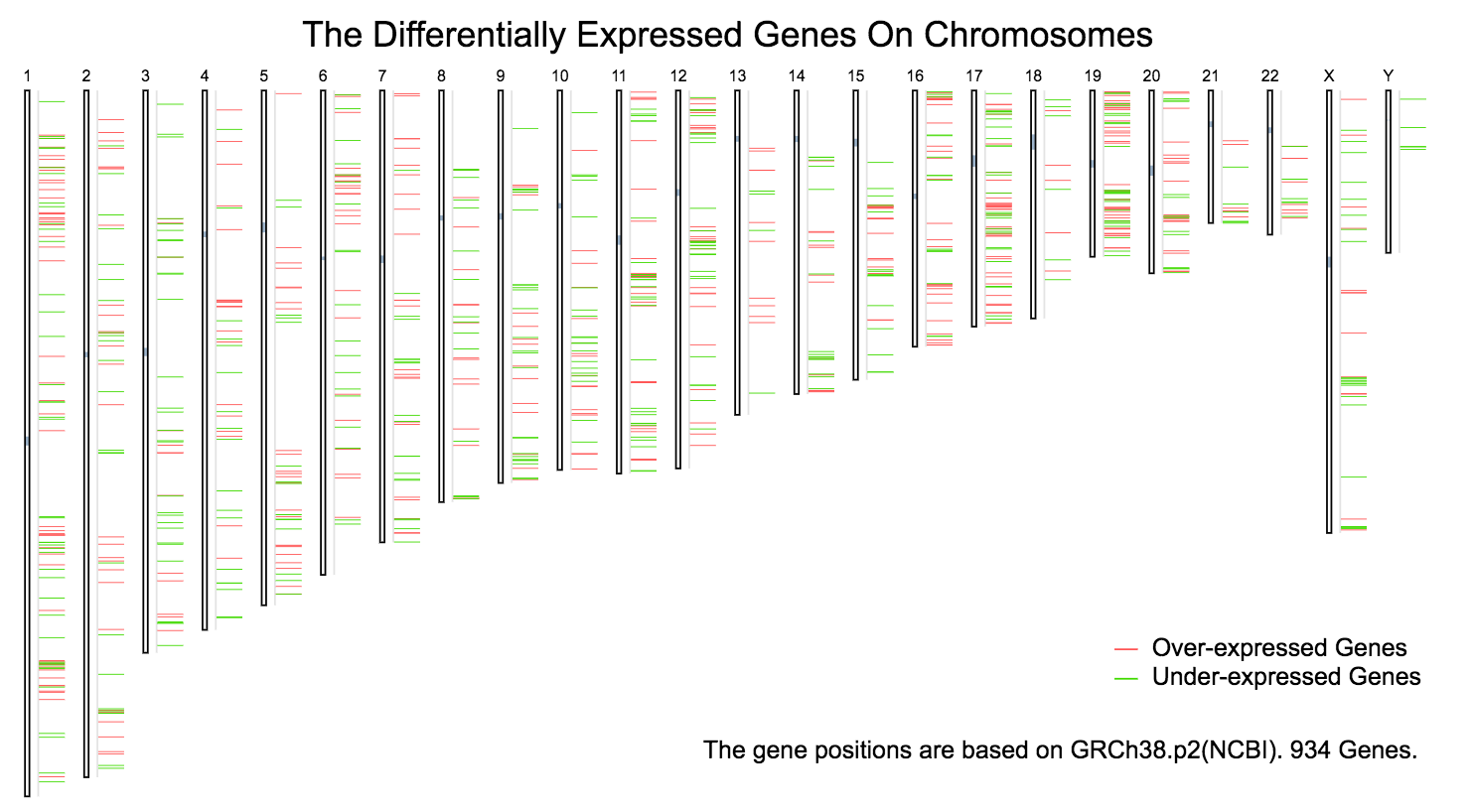
The chromosomal distribution of over- or under- expressed genes can be plotted in Differential Genes.
Over-expressed genes:
Under-expressed genes:
Both over-expressed and under-expressed genes:
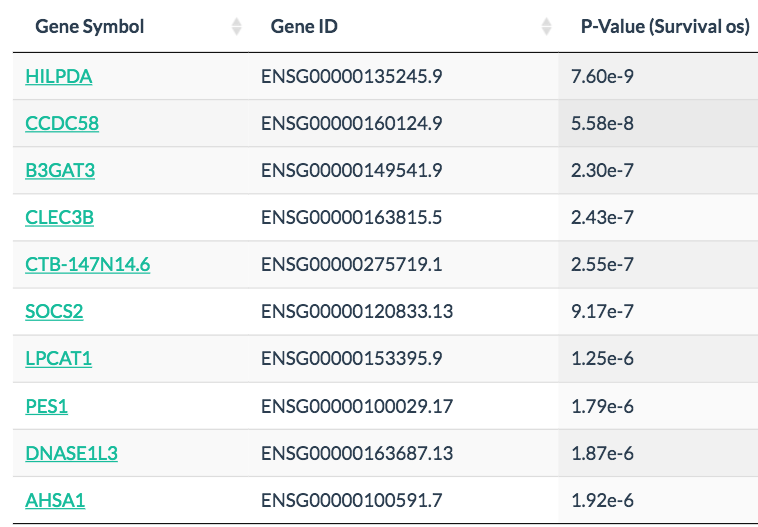
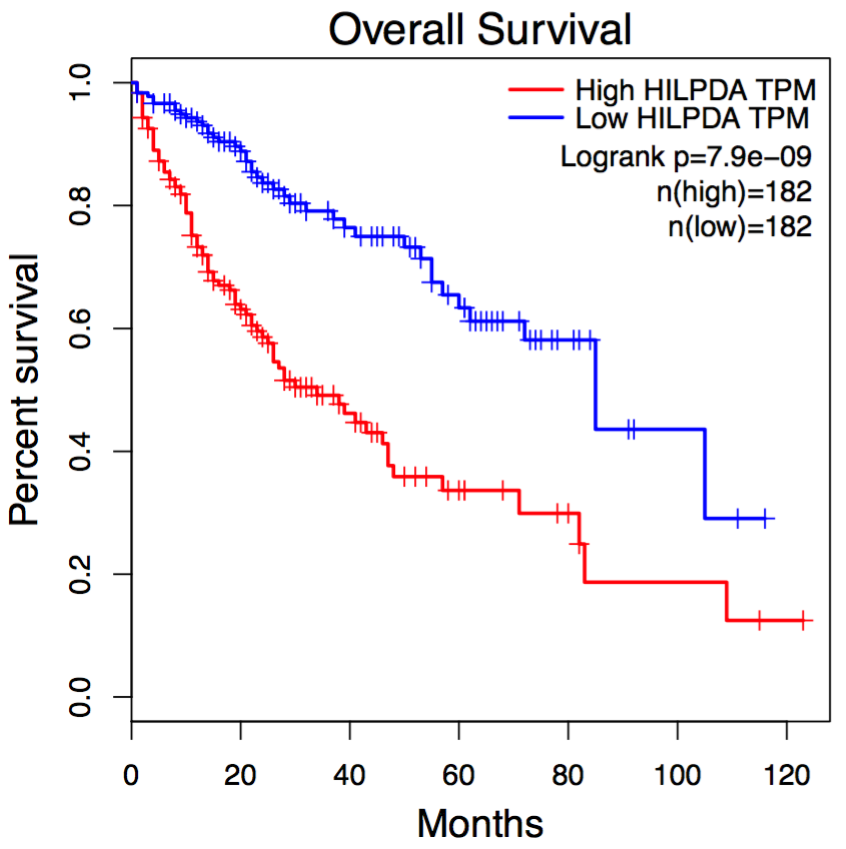
In Survival analysis, genes with the most significant association with patient survival can be identified, such as MCTS1 in breast cancer and HILPDA in liver cancer. Code
#!/usr/bin/Rscript
### input the parameters and connect to mysql database;
killDbConnections <- function () {
all_cons <- dbListConnections(MySQL())
for(con in all_cons){dbDisconnect(con)}
}
args<-commandArgs(T)
signature = args[1] ## signature genelist
dataset = args[2] ## selected datasets
parameter = args[3] ## parameters
signatures = strsplit(signature,",")[[1]] ## split the signature genelist into array
datasets = strsplit(dataset,",")[[1]] ## split the datasets info into array
parameters = strsplit(parameter,",")[[1]]
method = parameters[1] ## OS or DFS
symbol = parameters[2]
symbol2 = parameters[3]
ifreverse = parameters[4]
ifmonth = parameters[5]
highcutoff = parameters[6]
lowcutoff = parameters[7]
ifhr = parameters[8]
ifconf = parameters[9]
outputdir = parameters[10] ## outputdir
if(is.na(signatures[2])){signature = signatures[1]}else{
signature = paste(signatures,collapse = "','")
symbol = paste(symbol,symbol2,sep = "/")
}
dbs = "--------" ## database
.libPaths(c("--------",.libPaths())) ## add the specific directory of RMySQL R package
suppressPackageStartupMessages(library("RMySQL"))
killDbConnections()
mydb = dbConnect(MySQL(), user='root', password='--------$', dbname=dbs) ## connet to mysql database
df = as.matrix(array(data = 0,dim = c(0,length(signatures))))
for(table_t in datasets){
# table = table_t
if(table_t == ""){next}
table = paste(table_t,"_Tumor",sep = "")
df_t=t(dbGetQuery(mydb,paste("SELECT * FROM ",table," WHERE geneid IN ('",signature,"')",sep="")))
colnames(df_t) = df_t[1,]
df_t = df_t[-1,,drop = F]
df = rbind(df,df_t)
}
df = df[,signatures,drop=F]
storage.mode(df) = "numeric"
if(!is.na(signatures[2])){
df = log2(df + 0.001)
df = df[,1,drop = F] - df[,2,drop = F]
}
rownames(df) = sub(pattern="(.*_.*_.*)_.*$", replacement="\\1", rownames(df))
rownames(df) = chartr("_","-",rownames(df))
dataset_cut = gsub(pattern="(.*?)_.*$", replacement="\\1", datasets,perl = T)
dbs = "--------" ## database
.libPaths(c("/data/tangzefang/lib/R",.libPaths())) ## add the specific directory of RMySQL R package
suppressPackageStartupMessages(library("RMySQL"))
mydb = dbConnect(MySQL(), user='root', password='--------$', dbname=dbs) ## connet to mysql database
df_sur_t=(dbGetQuery(mydb,paste("SELECT * FROM survival WHERE CANCER IN ('",paste(dataset_cut,collapse = "','"),"')",sep="")))
df_sur_t = df_sur_t[df_sur_t[,"OSDAY"] != 0,]
df = df[rownames(df)[rownames(df) %in% df_sur_t[,1]],,drop = F]
high_df = rownames(df[df[,1] > quantile(df[,1],as.numeric(highcutoff)/100),1,drop = F])
low_df = rownames(df[df[,1] < quantile(df[,1],as.numeric(lowcutoff)/100),1,drop = F])
high_num = length(high_df)
low_num = length(low_df)
if(method == "dfs"){
df_sur = df_sur_t[df_sur_t[,1] %in% c(high_df,low_df),c(1:4)]
df_sur = cbind(df_sur,0)
df_sur[df_sur[,4] != "-",2] = 2 ## 2 means event while 1 means censor
df_sur[df_sur[,4] != "-",3] = df_sur[df_sur[,4] != "-",4]
df_sur[df_sur[,4] == "-",2] = 1
df_sur[df_sur[,2] == "Dead",2] = 2
df_sur = df_sur[,c(1,2,3,5)]
title = "Disease Free Survival"
}else{
df_sur = df_sur_t[df_sur_t[,1] %in% c(high_df,low_df),1:3]
df_sur = cbind(df_sur,0)
df_sur[df_sur[,2] != "Dead",2] = 1 ## 2 means event while 1 means censor
df_sur[df_sur[,2] == "Dead",2] = 2
title = "Overall Survival"
}
if(ifmonth == "month"){df_sur[,3] = as.numeric(df_sur[,3]) %/% 30 + 1}
df_sur[df_sur[,1] %in% high_df,4] = "high"
df_sur[df_sur[,1] %in% low_df,4] = "low"
df_sur = as.data.frame(df_sur)
df_sur[,3] = as.numeric(as.vector(df_sur[,3]))
df_sur[,2] = as.numeric(as.vector(df_sur[,2]))
colnames(df_sur)[4] = "CLASS"
#### modified code
df_sur$CLASS = factor(df_sur$CLASS,levels = c("low","high"))
if(!ifreverse == "reverse"){color = c(4,2)}else{color = c(2,4)}
suppressPackageStartupMessages(library(survival))
mod = Surv(df_sur$OSDAY,df_sur$OSEVENT)
mfit = survfit(mod~df_sur$CLASS)
sur = survdiff(mod~df_sur$CLASS)
p.val <- 1 - pchisq(sur$chisq, length(sur$n) - 1)
p.val = signif(p.val,2)
pdf(outputdir,width = 5,height = 5)
par(mar = c(4,4,2,1))
if(ifconf == "conf"){plot(mfit,col = color, lwd = c(2,1.3,1.3,2,1.3,1.3),mark.time=T,conf.int = T,lty = c(1,3,3,1,3,3))}else{plot(mfit,col = color, lwd = 2,mark.time=T)}
results_coxph = summary(coxph(mod~df_sur$CLASS))$coefficients
results_coxph_class = sub(pattern="df_sur\\$CLASS", replacement="", rownames(results_coxph))
results_coxph_hr = signif(results_coxph[1,"exp(coef)"],2)
results_coxph_hr_p = signif(results_coxph[1,"Pr(>|z|)"],2)
title(main = title,cex.main = 1.5,font.main = 1,line = 0.4)
if(ifmonth == "month"){xlab = "Months"}else{xlab = "Days"}
title(xlab = xlab,ylab="Percent survival",cex.lab=1.3,line = 2.5)
legend("topright",c(paste("Low",symbol,"TPM",sep=" "),
paste("High",symbol,"TPM",sep=" ")),
col = color,lty = 1 ,lwd = 2,bty = "n",xjust = 1,
y.intersp = 0.8,x.intersp = 0.5)
if(ifhr == "hr"){text(x = max(df_sur$OSDAY) * 1.04,y = 0.73,
labels = paste("Logrank p=",p.val,
"\n HR(",results_coxph_class,")=",results_coxph_hr,
"\n p(HR)=",results_coxph_hr_p,
"\n n(high)=",high_num,"\nn(low)=",low_num,
sep=""),pos = 2)
}else{text(x = max(df_sur$OSDAY) * 1.04,y = 0.8,labels = paste("Logrank p=",p.val,"\n n(high)=",high_num,"\nn(low)=",low_num,sep=""),pos = 2)}
garbage <- dev.off()
MCTS1 in breast cancer
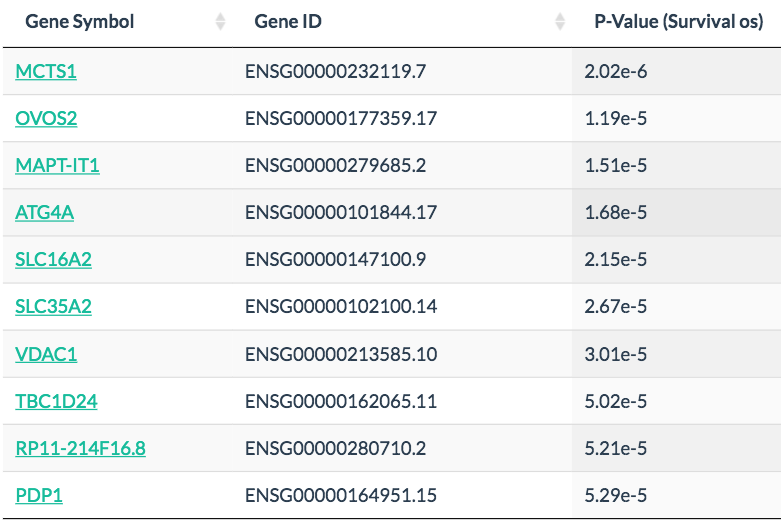
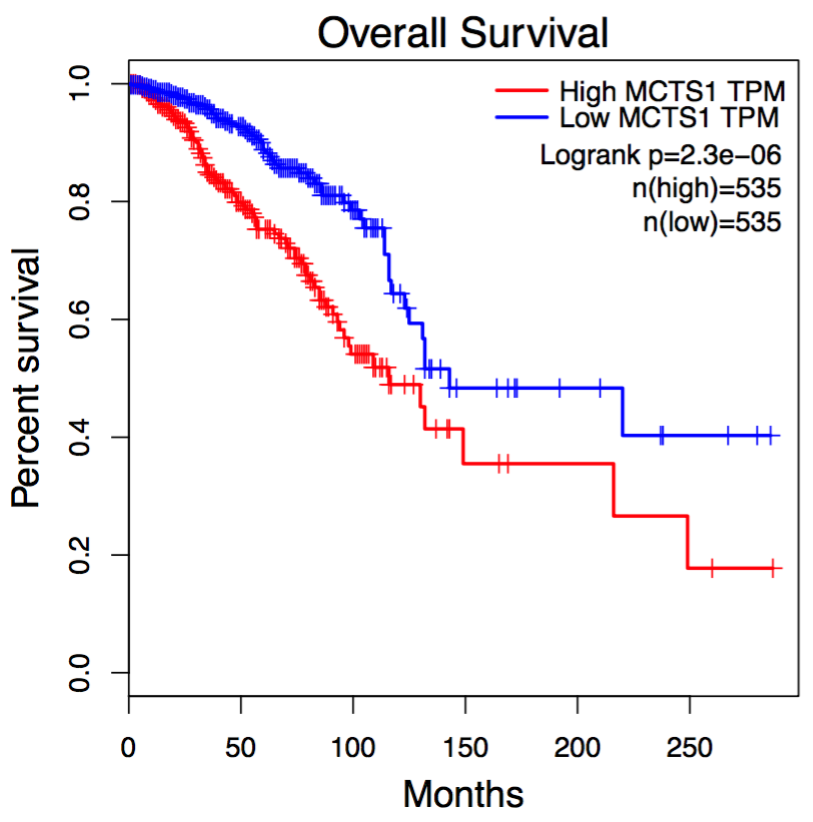
HILPDA in liver cancer:
Gene expression is visualized by both a bodymap and a bar plot in General.
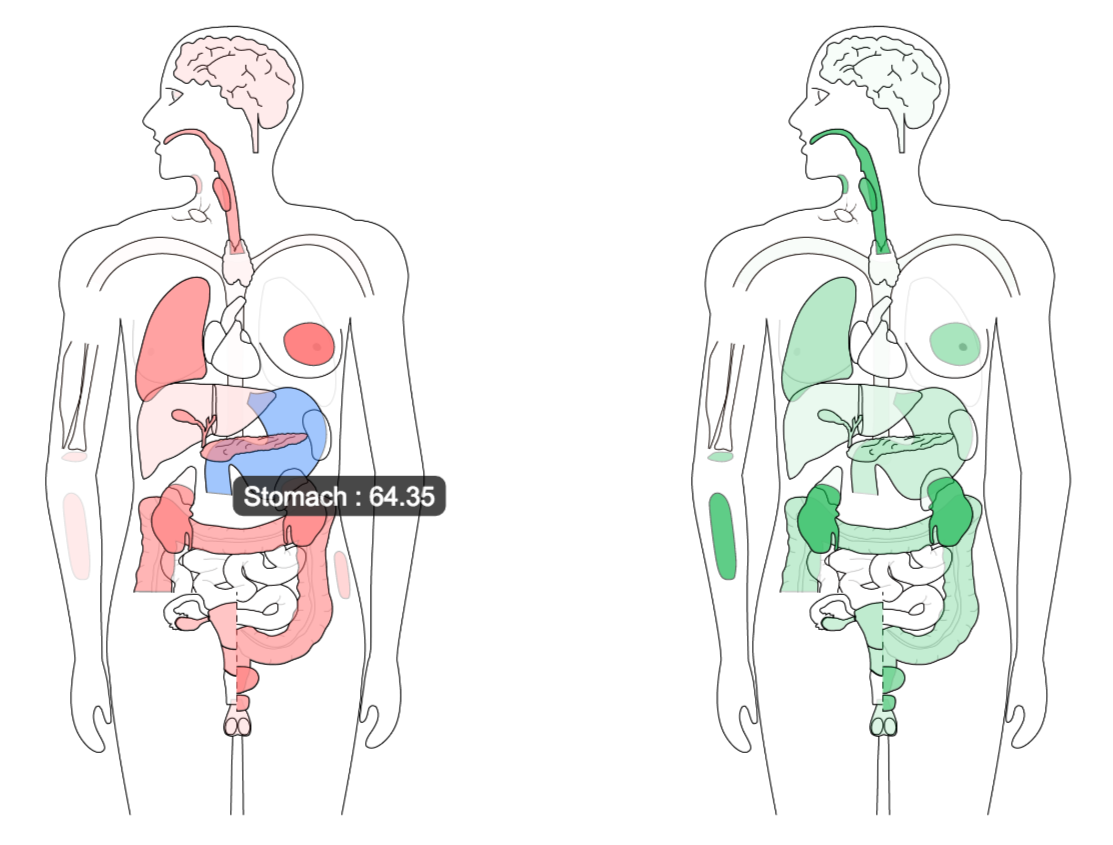
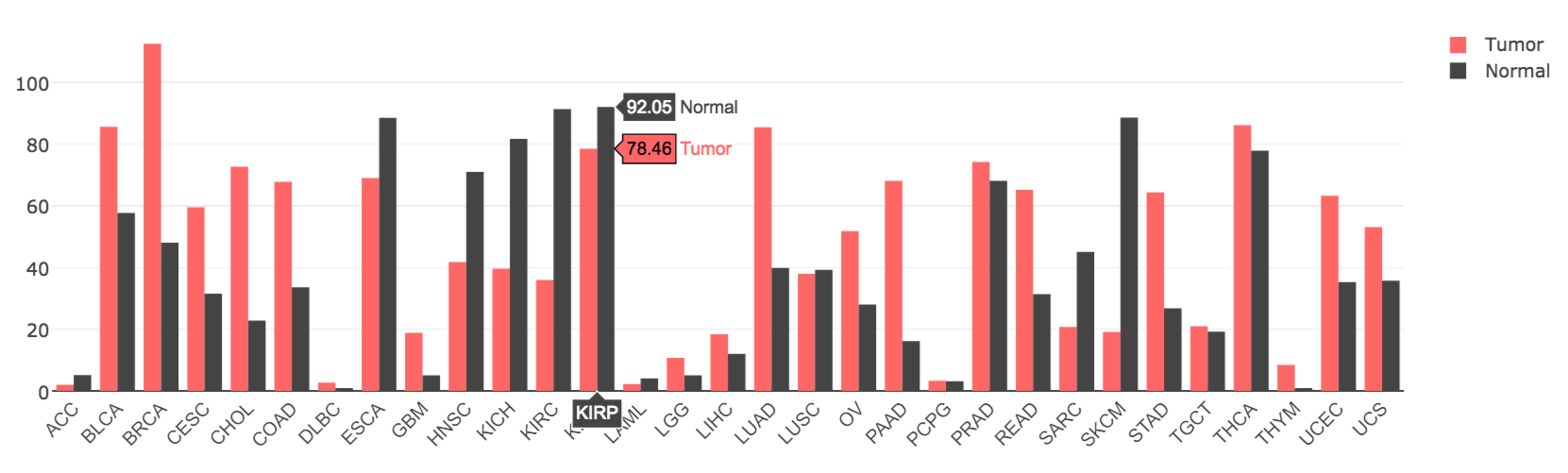
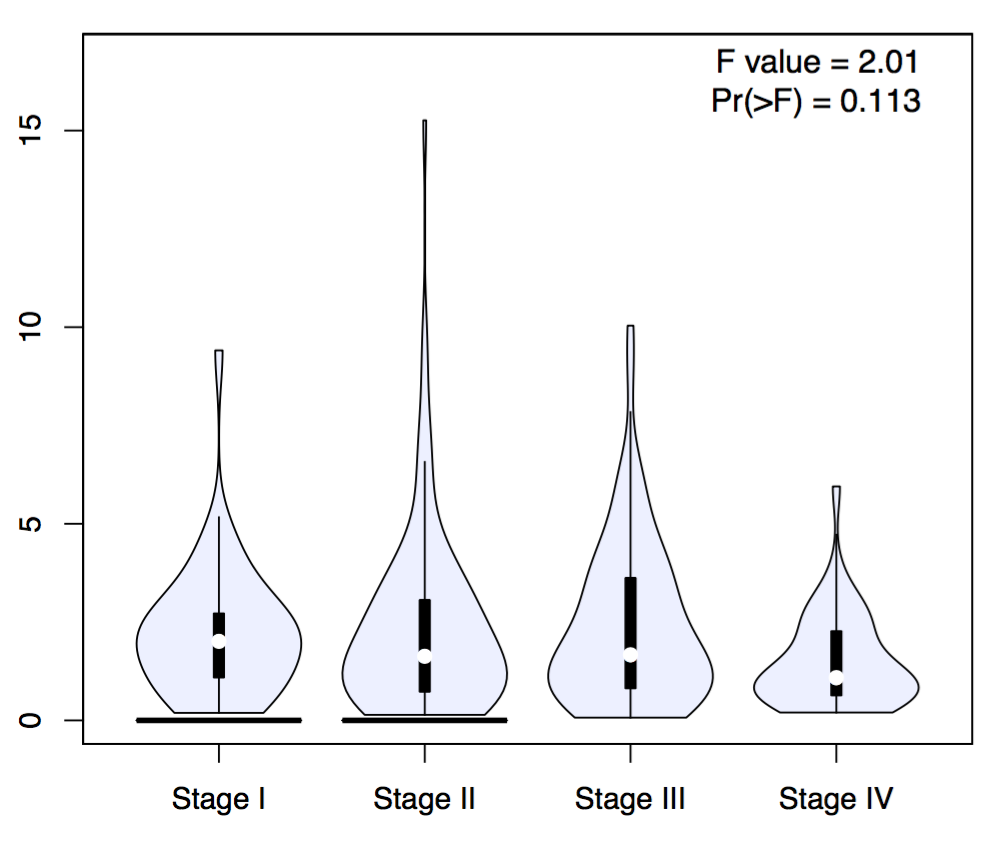
Gene expression by pathological stage is plotted in Stage plot. Code
#!/usr/bin/Rscript
killDbConnections = function () {
all_cons = dbListConnections(MySQL())
for(con in all_cons){dbDisconnect(con)}
}
args=commandArgs(T)
signature = args[1] ## signature genelist
dataset = args[2] ## selected datasets
parameter = args[3] ## parameters
parameters = strsplit(parameter,",")[[1]]
datasets = strsplit(dataset,",")[[1]]
datasets = datasets[datasets != ""]
ifmajor = parameters[1] ## iflog ["yes" or "no"]
iflog = parameters[2]
color = parameters[3]
outputdir = parameters[4] ## outputdir
if(ifmajor == "yes"){table = "stage_major";seq = c("Stage 0","Stage I","Stage II","Stage III","Stage IV","Stage X");}else{
table = "stage";seq = c("Stage 0","Stage I","Stage IA","Stage IA1","Stage IA2","Stage IB","Stage IB1","Stage IB2","Stage IC","Stage IS","Stage II","Stage IIA","Stage IIA1","Stage IIA2","Stage IIB","Stage IIC","Stage III","Stage IIIA","Stage IIIB","Stage IIIC","Stage IIIC1","Stage IIIC2","Stage IV","Stage IVA","Stage IVB","Stage IVC","Stage X");
}
dbs = "GE_SF"
.libPaths(c("--------",.libPaths())) ## add the specific directory of RMySQL R package
suppressPackageStartupMessages(library("RMySQL"))
suppressPackageStartupMessages(library("vioplot"))
killDbConnections()
#mydb = dbConnect(MySQL(), user='--------', password='--------', dbname=dbs) ## connet to mysql database
mydb = dbConnect(MySQL(), user='--------', password='--------', dbname=dbs,unix.socket="--------")
df = as.matrix(array(data = 0,dim = c(0,length(signature))))
for(table_t in datasets){
if(table_t == ""){next}
table_t = paste(table_t,"_Tumor",sep = "")
df_t=t(dbGetQuery(mydb,paste("SELECT * FROM ",table_t," WHERE geneid IN ('",signature,"')",sep="")))
colnames(df_t) = df_t[1,]
df_t = df_t[-1,,drop = F]
df = rbind(df,df_t)
}
# df = df[,signatures]
storage.mode(df) = "numeric"
if(iflog == "yes"){df = log2(df + 1)}
rownames(df) = sub(pattern="(.*_.*_.*)_.*$", replacement="\\1", rownames(df))
rownames(df) = chartr("_","-",rownames(df))
dataset_cut = gsub(pattern="(.*?)_.*$", replacement="\\1", datasets,perl = T)
dbs = "GE_STAGE" ## database
.libPaths(c("--------",.libPaths())) ## add the specific directory of RMySQL R package
suppressPackageStartupMessages(library("RMySQL"))
#mydb = dbConnect(MySQL(), user='--------', password='--------', dbname=dbs) ## connet to mysql database
mydb = dbConnect(MySQL(), user='--------', password='--------', dbname=dbs,unix.socket="--------")
df_stage=(dbGetQuery(mydb,paste("SELECT * FROM ",table," WHERE CANCER IN ('",paste(dataset_cut,collapse = "','"),"')",sep="")))
colnames(df_stage)[3] = "value"
rownames(df_stage) = df_stage[,1]
df_stage = df_stage[,-1]
df_stage = df_stage[rownames(df_stage)[rownames(df_stage) %in% rownames(df)],,drop = F]
df_stage[,2] = df[rownames(df_stage),1]
list_stage = table(df_stage[,1])
if(length(list_stage[list_stage == 1]) > 0){df_stage = df_stage[df_stage[,1] != names(list_stage[list_stage == 1]),]}
seq_certain = seq[seq %in% df_stage[,1]]
n = length(unique(df_stage[,1]))
dist = max(range(df_stage[,2]))/10
ylim = range(df_stage[,2])
ylim[2] = ylim[2]+dist
pdf(outputdir,title="Result Display",width = 2 + n,height = 5)
par(mar=c(3.6, 3.1, 1.6, 2.1))
boxplot(0,0 , xlim=c(0.5,n + 0.5), ylim=ylim, xaxt="n", col="yellow")
for(i in 1:n){
vioplot(x = df_stage[df_stage[,1] == seq_certain[i],2],at = i,add = T,col = color)
}
if(iflog == "yes"){aov_model = aov(value ~ STAGE,data = df_stage)}else{
df_stage_log = df_stage
df_stage_log[,2] = log2(df_stage_log[,2] + 1)
aov_model = aov(value ~ STAGE,data = df_stage_log)
}
stat_result = summary(aov_model)[[1]][1,][4:5]
text(x = n + 0.5 ,y=max(df_stage[,2])*1.05,labels = paste("F value = ",signif(stat_result[1,1],3),"\nPr(>F) = ",
signif(stat_result[1,2],3),sep=""),cex = 1.1,col = "black",pos = 2)
axis(1, at = 1:n, labels = seq_certain, tick = TRUE)
blackhole = dev.off()
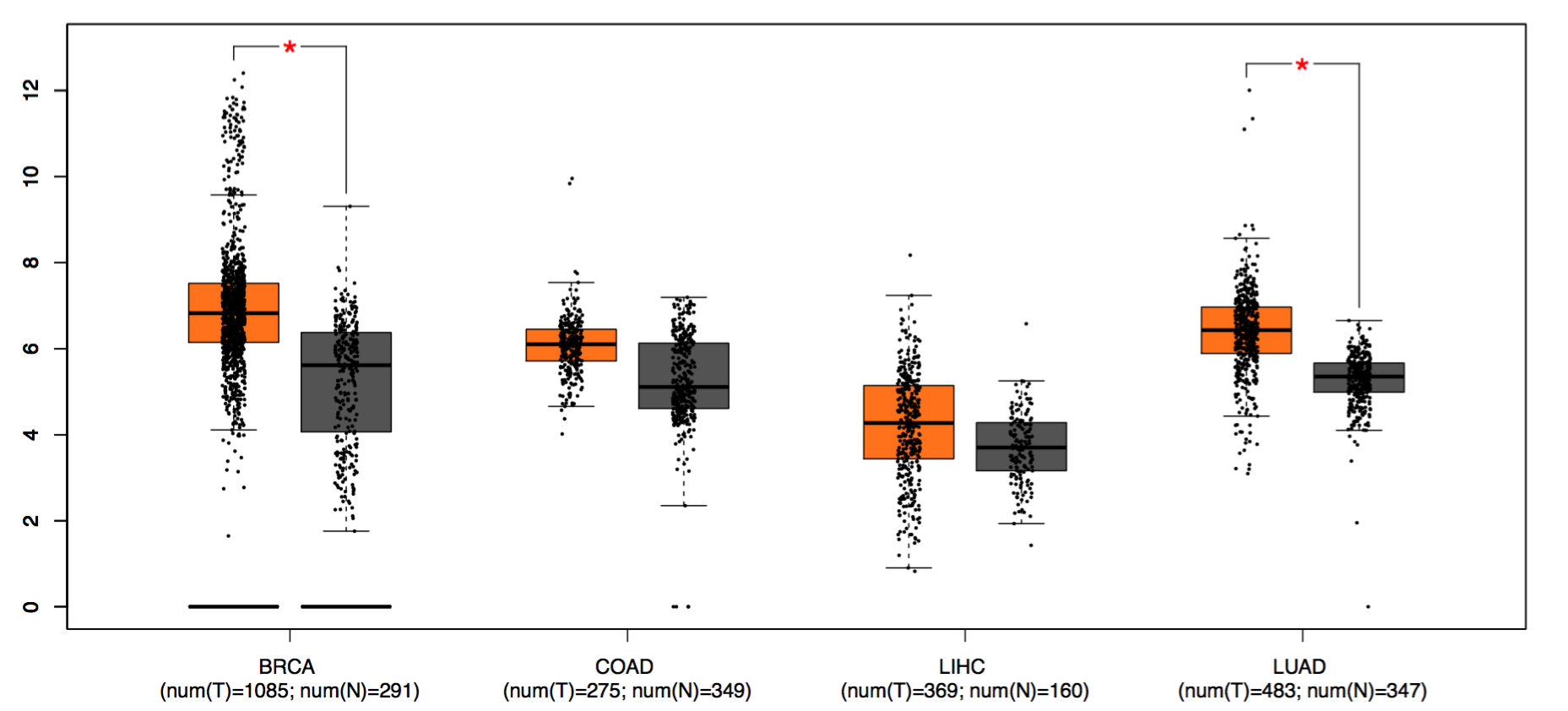
Users can compare the expression of one gene in multiple cancers by Boxplot, or compare multiple genes by a matrix plot in Multiple gene comparison. Code
#!/usr/bin/Rscript
args=commandArgs(T)
signature = args[1] ## signature genelist
dataset = args[2] ## selected datasets
parameter = args[3] ## parameters
datasets = strsplit(dataset,",")[[1]] ## split the datasets info into array
datasets = datasets[datasets != ""]
parameters = strsplit(parameter,",")[[1]]
dbs = "GE_SF" ## database
iflog = parameters[1] ## iflog ["yes" or "no"]
suffix = parameters[2] ## "NG" or "Normal"
color1 = parameters[3]
color2 = parameters[4]
fccutoff = as.numeric(parameters[5])
qcutoff = as.numeric(parameters[6])
jittersize = as.numeric(parameters[7])
outputdir = parameters[8] ## outputdir
.libPaths(c("--------",.libPaths())) ## add the specific directory of RMySQL R package
suppressPackageStartupMessages(library("RMySQL"))
#mydb = dbConnect(MySQL(), user='--------', password='--------', dbname=dbs) ## connet to mysql database
mydb = dbConnect(MySQL(), user='--------', password='--------', dbname=dbs,unix.socket="--------")
### build the integrated table
### single gene
df = as.matrix(array(data = 0,dim = c(0,2)))
for(j in c("Tumor",suffix)){
for(i in 1:length(datasets)){
if(datasets[i] == ""){next;}
table = paste(datasets[i],j,sep="_")
df_t=t(dbGetQuery(mydb,paste("SELECT * FROM ",table," WHERE geneid IN ('",signature,"')",sep="")))
colnames(df_t) = df_t[1,]
df_t = df_t[-1,,drop = F]
df_t = cbind(df_t,table)
df = rbind(df,df_t)
}
}
colnames(df)[2] = "class"
df = as.data.frame(df)
df[,1] = as.numeric(as.vector(df[,1]))
if(iflog == "yes"){
df[,1]=log2(df[,1]+1)
}
n = length(datasets)
## hypothesis test
stat_df = as.matrix(array(data = 0,dim = c(2,n)))
colnames(stat_df) = datasets
rownames(stat_df) = c("LogFC","pvalue")
mark = rep(NA,n)
for(i in 1:length(datasets)){
if(iflog != "yes"){df_t = df;df_t[,1] = log2(df_t[,1]+1);}else{df_t = df;}
colnames(df_t) = c("value","class")
df_t_tumor = df_t[df_t[,2] == paste(datasets[i],"_Tumor",sep=""),]
df_t_normal = df_t[df_t[,2] == paste(datasets[i],"_",suffix,sep=""),]
stat_df["LogFC",i] = as.numeric(median(df_t_tumor[,1]) - median(df_t_normal[,1]))
df_t_both = rbind(df_t_tumor,df_t_normal)
aov_model = aov(value ~ class,data = df_t_both)
stat_df["pvalue",i]=as.numeric(summary(aov_model)[[1]][1,][5])
if(abs(stat_df["LogFC",i]) > fccutoff & stat_df["pvalue",i] > qcutoff){mark[i]= "*";}else{mark[i] = NA;}
}
### boxplot function
pdf(outputdir,title="Result Display",width = 1+3*n,height = 6)
par(mar=c(3.6, 3.1, 1.6, 2.1))
dist = max(range(df[,1]))/20
ylim = range(df[,1])
ylim[2] = ylim[2]+dist
boxplot(0,0,xlim=c(0,n*3), ylim=ylim, xaxt="n", col="red",cex = 0.5,outline=F)
for(i in 1:length(datasets)){
seq_t = c(paste(datasets[i],"_Tumor",sep=""),paste(datasets[i],"_",suffix,sep=""))
df_t = df[df[,2] %in% seq_t,]
df_t[,2] = factor(as.vector(df_t[,2]),levels = seq_t)
colnames(df_t) = c("value","class")
boxplot(value~class,data = df_t, at=c((i-1)*3+1,(i-1)*3+2), xaxt="n", add=TRUE, col=c(color1,color2),cex = 0.5,outline=F)
stripchart(value~class, vertical = TRUE, data = df_t,at=c((i-1)*3+1,(i-1)*3+2),
method = "jitter", add = TRUE, pch = 20, col = 'black',cex = jittersize)
}
xpos = 0:(n-1)*3+1.5
ypos.a = c()
for(i in paste(datasets,"Tumor",sep="_")){
ypos.a = c(ypos.a,max(df[df[,2] == i,1]))
}
ypos.b = c()
for(i in paste(datasets,suffix,sep="_")){
ypos.b = c(ypos.b,max(df[df[,2] == i,1]))
}
for(i in 1:length(mark)){
if(!is.na(mark[i])){
segments(xpos[i]-.5, ypos.a[i]+dist/2, xpos[i]-.5, max(ypos.a[i], ypos.b[i])+dist)
segments(xpos[i]+.5, ypos.b[i]+dist/2, xpos[i]+.5, max(ypos.a[i], ypos.b[i])+dist)
segments(xpos[i]-.5, max(ypos.a[i], ypos.b[i])+dist, xpos[i]-0.1, max(ypos.a[i], ypos.b[i])+dist)
segments(xpos[i]+.5, max(ypos.a[i], ypos.b[i])+dist, xpos[i]+0.1, max(ypos.a[i], ypos.b[i])+dist)
text(x=xpos[i], y=max(ypos.a[i], ypos.b[i])+dist, label=mark[i], col="red",cex = 2)
}
}
label=c()
for(i in 1:length(datasets)){
label_t_num = sum(df[,2] == paste(datasets[i],"Tumor",sep="_"))
label_n_num = sum(df[,2] == paste(datasets[i],suffix,sep="_"))
label_t = paste(datasets[i]," \n(num(T)=",label_t_num,"; num(N)=",
label_n_num,")",sep="")
label = c(label,label_t)
}
axis(1, at = 0:(n-1)*3+1.5,mgp=c(2,2,0), labels = label, tick = TRUE)
a = dev.off()
Boxplot:
Matrix plot:
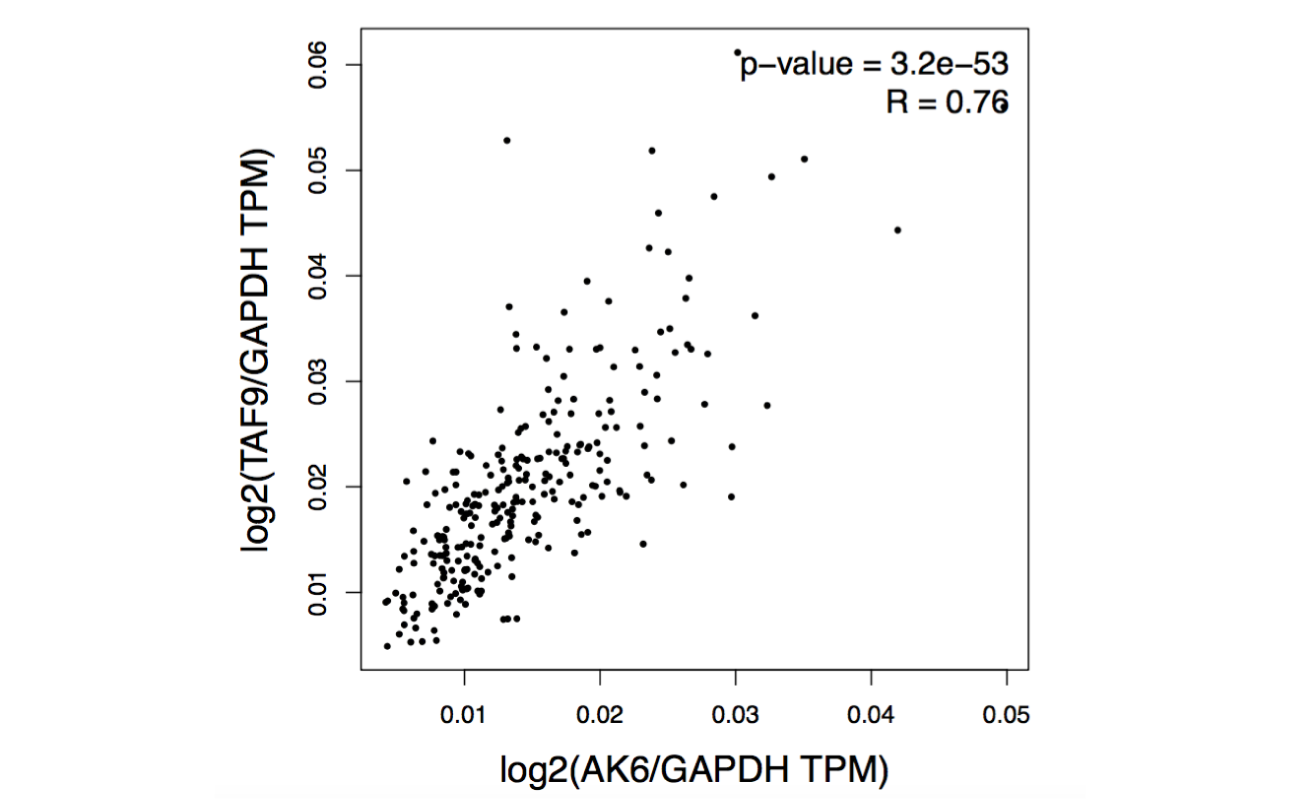
GEPIA provides pair-wise gene correlation analysis of a given set of TCGA and/or GTEx expression data. Normalization is optional and customizable. Code
#!/usr/bin/Rscript
killDbConnections = function () {
all_cons = dbListConnections(MySQL())
for(con in all_cons){dbDisconnect(con)}
}
args=commandArgs(T)
signature = args[1] ## signature genelist
dataset = args[2] ## selected datasets
parameter = args[3] ## parameters
signatures = strsplit(signature,",")[[1]] ## split the signature genelist into array
if(is.na(signatures[4])){signatures[4] = ""}
datasets = strsplit(dataset,",")[[1]] ## split the datasets info into array
datasets = datasets[datasets != ""]
parameters = strsplit(parameter,",")[[1]]
dbs = "GE_SF" ## database
symbol1 = parameters[1]
symbol2 = parameters[2]
symbol3 = parameters[3]
symbol4 = parameters[4]
method = parameters[5] ## "pearson" or "spearman" or "kendall"
outputdir = parameters[6] ## outputdir
.libPaths(c("--------",.libPaths())) ## add the specific directory of RMySQL R package
suppressPackageStartupMessages(library("RMySQL"))
killDbConnections()
#mydb = dbConnect(MySQL(), user='--------', password='--------', dbname=dbs) ## connet to mysql database
mydb = dbConnect(MySQL(), user='--------', password='--------', dbname=dbs,unix.socket="--------")
## Gene A:
if(signatures[3] != ""){
symbol1 = paste(symbol1,symbol3,sep = "/")
signatures_a = c(signatures[1],signatures[3])
}else{signatures_a = signatures[1]}
df_a = as.matrix(array(data = 0,dim = c(0,length(signatures_a))))
for(i in 1:length(datasets)){
table = datasets[i]
df_t=t(dbGetQuery(mydb,paste("SELECT * FROM ",table," WHERE geneid IN ('",paste(signatures_a, collapse = "','"),"')",sep="")))
colnames(df_t) = df_t[1,]
df_t = df_t[-1,,drop = F]
df_a = rbind(df_a,df_t)
}
storage.mode(df_a) = "numeric"
df_a = df_a[,signatures_a,drop = F]
if(signatures[3] != ""){
df_a = log2(df_a + 0.001)
df_a = df_a[,1,drop = F] - df_a[,2,drop = F]
df_a = 2^df_a
colnames(df_a) = signatures_a[1]
}
## Gene B:
if(signatures[4] != ""){
symbol2 = paste(symbol2,symbol4,sep = "/")
signatures_b = c(signatures[2],signatures[4])
}else{signatures_b = signatures[2]}
df_b = as.matrix(array(data = 0,dim = c(0,length(signatures_b))))
for(i in 1:length(datasets)){
table = datasets[i]
df_t=t(dbGetQuery(mydb,paste("SELECT * FROM ",table," WHERE geneid IN ('",paste(signatures_b, collapse = "','"),"')",sep="")))
colnames(df_t) = df_t[1,]
df_t = df_t[-1,,drop = F]
df_b = rbind(df_b,df_t)
}
storage.mode(df_b) = "numeric"
df_b = df_b[,signatures_b,drop = F]
if(signatures[4] != "" ){
df_b = log2(df_b + 0.001)
df_b = df_b[,1,drop = F] - df_b[,2,drop = F]
df_b = 2^df_b
colnames(df_b) = signatures_b[1]
}
df = cbind(df_a,df_b)
### build the integrated table
### single gene
colnames(df)[colnames(df) == signatures[1]] = symbol1
colnames(df)[colnames(df) == signatures[2]] = symbol2
pdf(file = outputdir,title="Result Display",width = 6,height = 5.5)
par(mar=c(4.5, 5.1, 1.1, 2.1))
options(warn=-1)
rvalue = signif(cor(x = df[,1], y = df[,2],method = method),2)
cpvalue = signif(as.numeric(cor.test(x = df[,1], y = df[,2],method = method)[3]),2)
plot(x = log2(df[,1] + 1),y = log2(df[,2] + 1),main = NULL,cex.lab = 1.5,
xlab = paste("log2(",colnames(df)[1]," TPM)",sep = ""),
ylab = paste("log2(",colnames(df)[2]," TPM)",sep = ""), pch = 19,cex=0.5)
range_y = range(log2(df[,2] + 1))
text(x = max(log2(df[,1] + 1)) * 1.03,y = max(log2(df[,2] + 1)) - (range_y[2] - range_y[1])/15,labels = paste("p-value = ",as.character(cpvalue),"\nR = ",as.character(rvalue),sep=""),cex = 1.3,col = "black",pos = 2)
a = dev.off()
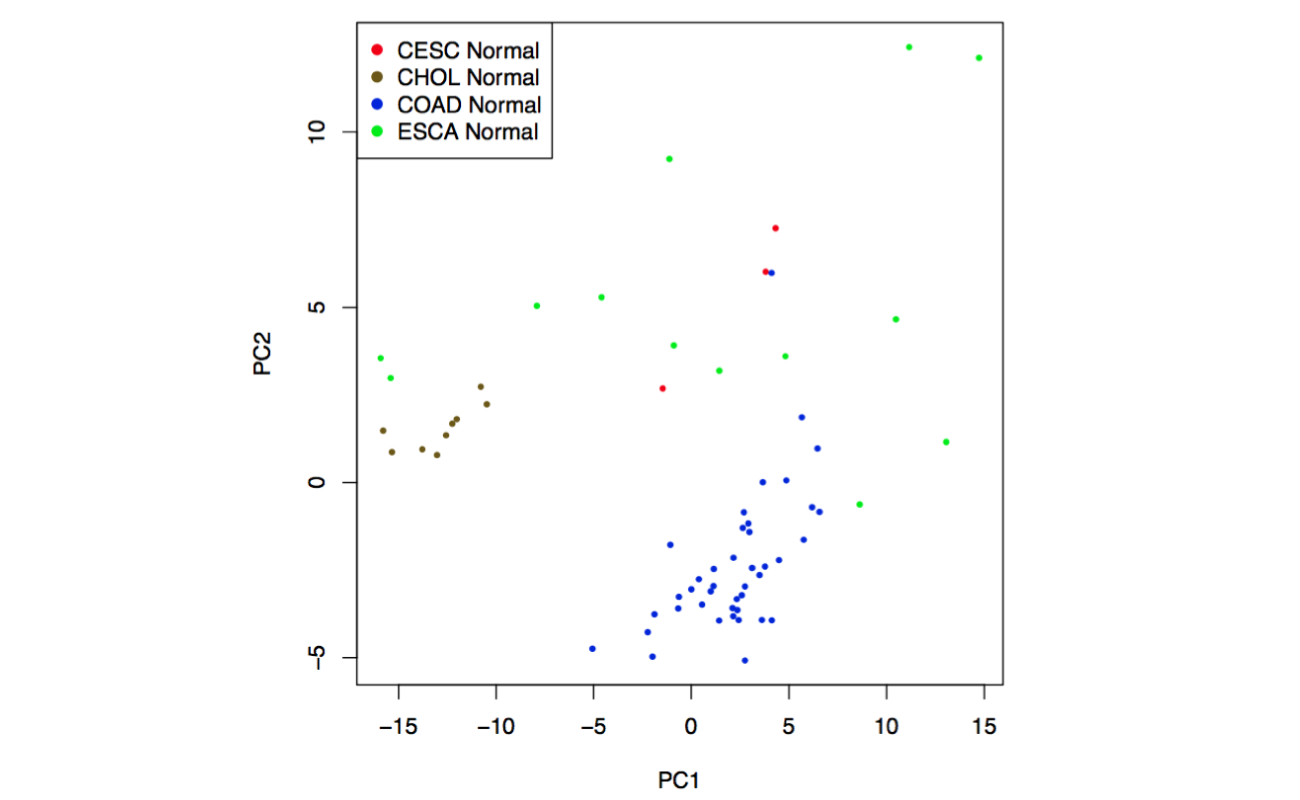
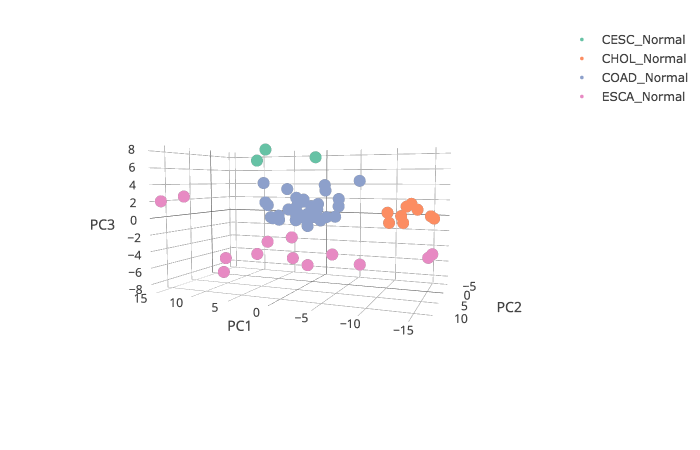
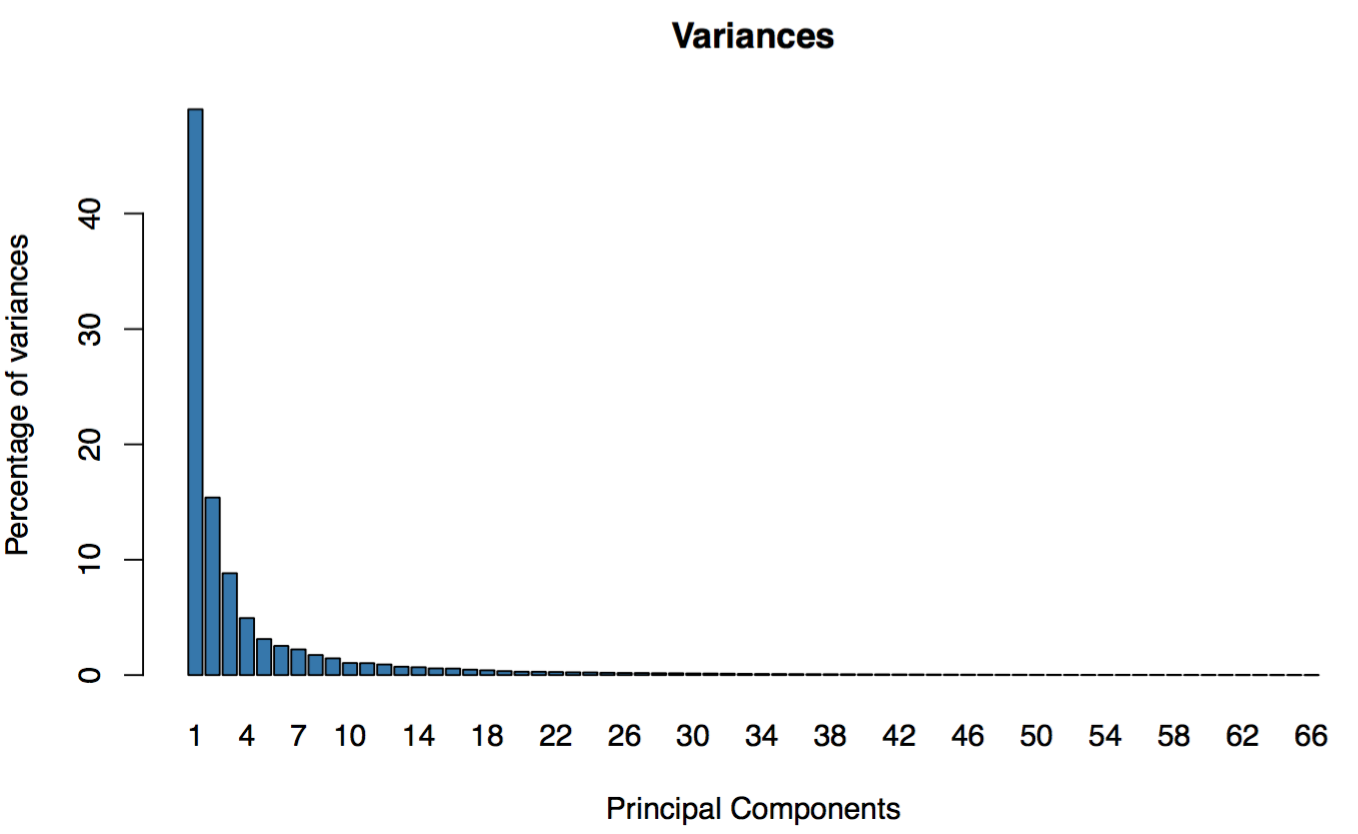
GEPIA provides Principal Component Analysis of multiple genes and cancer types in PCA, and presents results by 2D or 3D plots.
2D plots:
3D plots:
Variances distribution:
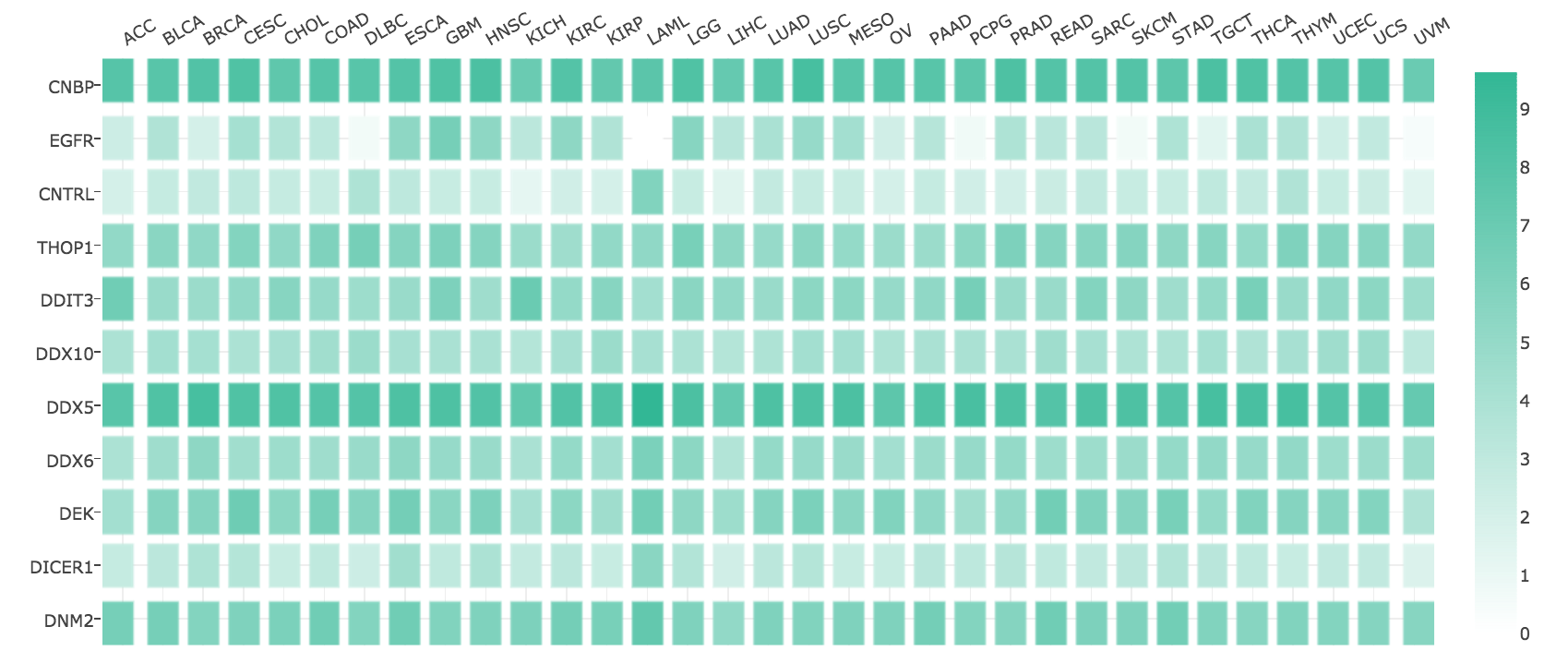
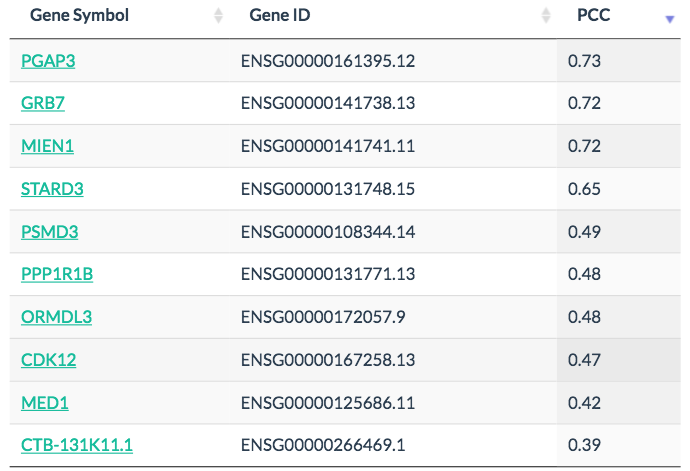
Genes with similar expression pattern can be identified in Similar Genes, for example, PGAP3 and GRB7 are similar to ERBB2.
ERBB2:
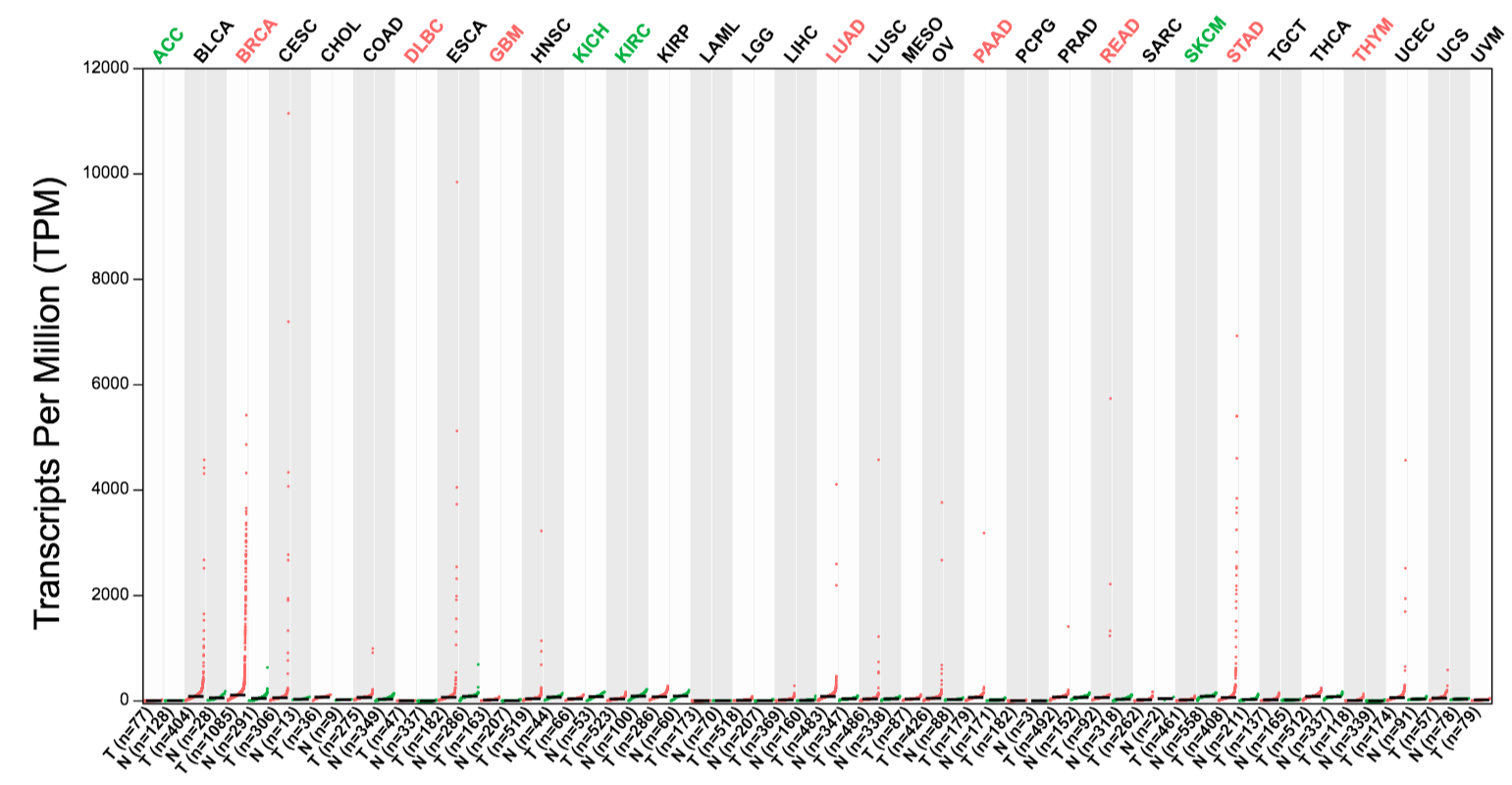
PGAP3:
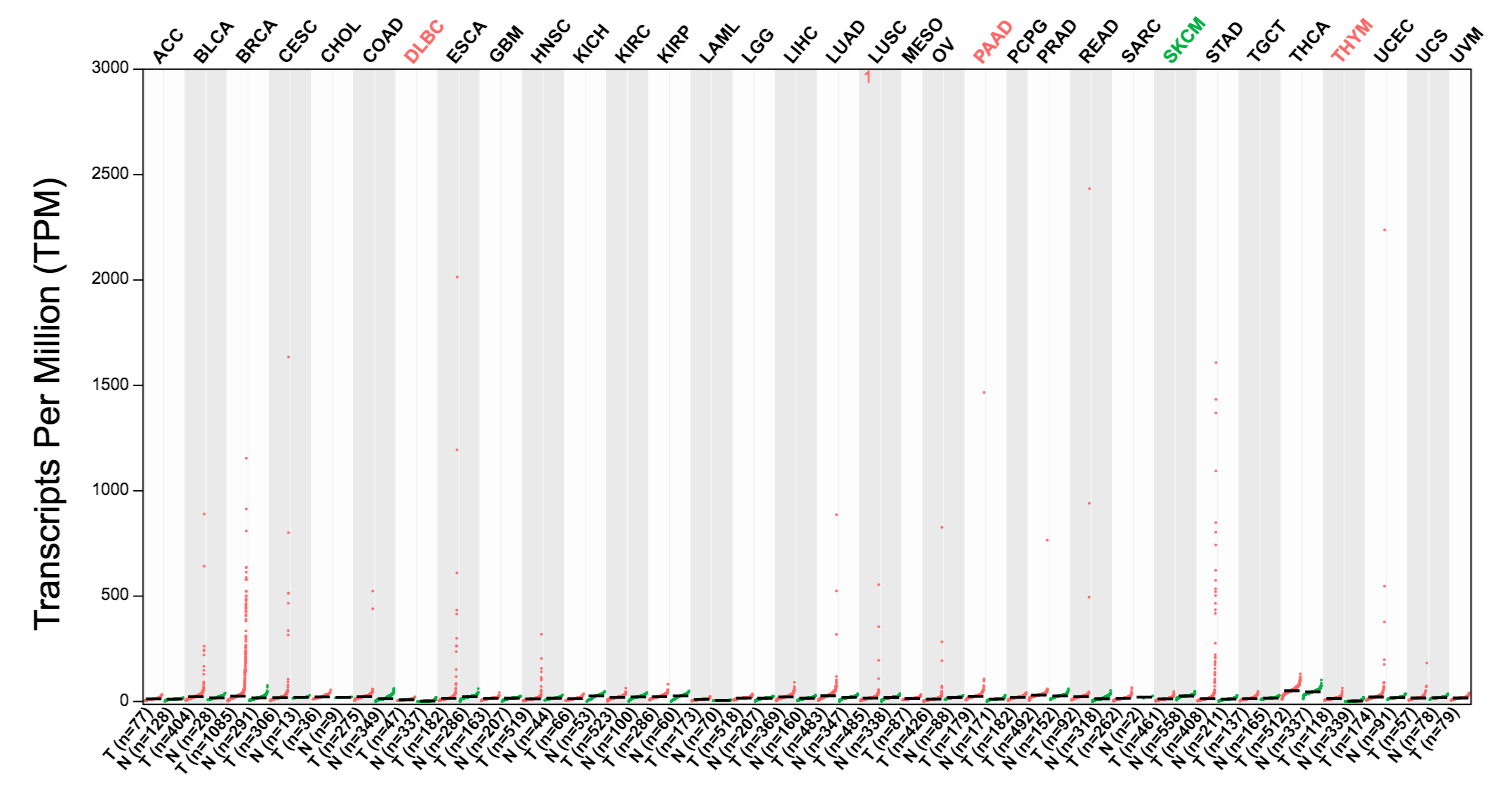
GRB7: